


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
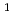 A' electronic states of H
A' electronic states of H
市原 晃; 横山 啓一; 岩本 修
JAERI-Data/Code 98-031, 18 Pages, 1998/11
 JAERI-Data-Code-98-031.pdf:0.89MB
JAERI-Data-Code-98-031.pdf:0.89MB
基底及び第一電子励起A'状態におけるHのポテンシャルエネルギーを701の異なる空間構造に対して計算し、その結果を表にまとめた。これらのポテンシャルエネルギーは、[8s6p2d1f]ガウス型基底関数を用いた、非経験的分子軌道論に基づく完全な配置間相互作用法により計算された。ポテンシャルの計算値を内挿することにより、二つのポテンシャル面間のavoided crossingや基底状態ポテンシャル面のエネルギー極小値付近の形状を表現することが可能である。ポテンシャルエネルギーは三つの核間距離の関数として0.6から10.0ボーアの範囲内で与えられており、H +H
+H 衝突に関する分子動力学の研究に適している。
衝突に関する分子動力学の研究に適している。
system with the trajectory-surface-hopping model市原 晃; 白井 稔三; 横山 啓一
Journal of Chemical Physics, 105(5), p.1857 - 1861, 1996/08
被引用回数:38 パーセンタイル:77.95(Chemistry, Physical)非経験的分子軌道計算(ab initio full configuration interaction calculation with a [8s6p2d1f] Gaussian type basis set)で得られたHの3次元ポテンシャル面上でtrajectory-surface-hopping法を用いることにより、D+H、D+D、及びH+D衝突で生じる電荷移動及び粒子組み替え反応の断面積を計算した。ポテンシャル面間の非断熱的電子遷移の確率は、Landau-Zener-Stuckelberg近似に基づいて評価した。その結果、衝突エネルギーが2.5eVから5.0eVの範囲では、計算結果は実験値と定量的に一致することが確認された。
A′ states of H市原 晃; 横山 啓一
Journal of Chemical Physics, 103(6), p.2109 - 2112, 1995/08
被引用回数:55 パーセンタイル:87.01(Chemistry, Physical)基底および第1電子励起(A′)状態におけるHの断熱ポテンシャルを、非経験的分子軌道論に基づき、full configuration interaction法を用いて計算した。水素原子の基底関数としては、ガウス型[8s6p2d1f]基底を用いた。Hの3次元ポテンシャルを得るために、680の異なる空間配置に対して計算を行っている。また、これらのポテンシャル間の非断熱的電子遷移の確率を、Landau-Zener-Stuckelberg近似により見積った。
